杜兴氏肌营养不良
更新日期:2019-09-21 07:41:45 来源:本站作者:admin杜兴氏肌营养不良是一种性联隐性遗传病,又名假肥大型肌营养不良症,是预后最严重的肌营养不良类型。治病先知病,什么是杜兴氏肌营养不良?
一、 概述:杜兴氏肌营养不良(Duchenne muscular dystrophy,简称DMD)是由于X染色体上dystrophin基因请缺陷所致,发病率为新生男婴的1/3500,该疾病的特点为肌肉进行性损伤,患者3-4岁左右发病,5-6岁可出现行走困难,临床表现为进行性加重的全身骨骼肌无力和肌肉萎缩。我国发病率为1/1000,多分布在山东、河南、河北、安徽、浙江等地,其中只有2/3是由于上代的X染色体病变造成,即遗传而来。故遗传造成的发病率是10-20/10000,其它1/3的发病不是由于上代,而是当代本身X染色体产生突变(缺失或移位)而造成,这一类患者虽不是遗传而来但对后代不遗传性。杜兴氏肌肉营养不良症最终会影响到所有平滑肌,以及心脏和呼吸肌肉。患者很少能活过35岁。 患者一般由于呼吸衰竭或心功能紊乱而死亡。
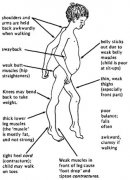
二、临床表现是:患儿均为男性,多在3-5岁发病;起病隐袭,大部分患儿坐、立时间发育正常,50%病儿开始行走较正常儿童晚(15个月后),开始症状多为行走慢,不能正常跑步,容易跌倒;肌无力自躯干和四肢近端开始缓慢进展,下肢重于上肢;骨盆带肌肉无力,肌张力减低,由于髂腰肌和股四头肌无力而登楼及蹲位站立因难,进而腰椎前突,因盆带肌无力而走路时向两侧摇摆,呈典型鸭步;由仰卧站立时由于股肌和髂腰肌的无力,患儿必须先转为俯卧位,然后以双手支撑双足背、膝部等处顺次露附,方能直立.称为GowerS征,为本病的特征性表现;肩胛带肌肉也同时受累,举臂无力,因前锯肌和斜方肌无力,不能固定肩胛内缘,使肩胛游离呈冀状支于背部,称为翼状肩胛,当双臂前推时尤为明显;一般四肢近端肌萎缩明显,双腓肠肌假性肥大见于90%患儿,是因萎缩肌纤维周围均被脂肪和结缔组织充填,故体积增大而肌力减弱,触之坚硬;假性肥大尚可见于臂肌、三角肌、冈下肌等;也可见轻度面肌无力,但发音、吞咽、眼肌运动不受累;由于病情进展,逐渐出现关节挛缩,常见于髋关节、膝关节、跟腱,后期出现肘关节及躯干肌挛缩。
一般至9-12岁患儿不能行走,要坐轮椅。多数患儿心肌受累,表现为窦性心动过速,异常的R波,V1导联S波变浅,深的Q波(胸导联),P-R间期缩短,V1导联出现Rsr波群及束支传导阻滞。晚期出现心脏扩大,心力衰竭,心律紊乱。约18-20岁时患者出现呼吸窘迫症状,晚期病情加重需呼吸机支持。大约有三分之一的DMD患者有一定程度的学习障碍,但一般不会有严重的愚钝。医生认为抗肌萎缩蛋白的不正常,使大脑中发生了微妙的变化,引起了认识和行为的障碍。DMD患者的智力问题主要表现在:注意力不容易集中、词汇学习和记忆较差、情感交流有一定困难。一般无消化道症状。存活很少超过30岁。本型的病情是肌营养不良中最严重的,其严重程度与患儿家族中遗传代数成反比.即家族中受累代数越多,病情越轻,严重的是散发病例.预后不良。
四、征兆及诊断方法:一般在5岁以前发病;临床特点为进行性对称性肌无力,以肢体近端受累多见,起病常于下肢开始;体查无肌颤,无感觉障碍,多伴有腓肠肌假性肥大;血清肌酸肌酶(CK)增高数十或数百倍;肌电图呈肌源性损害;肌活检表现为肌纤维长短不一,出现坏死与降解,纤维透明化,出现结缔组织与脂肪组织代偿增生,免疫组化分析可见dystrophin缺失;有家族史,呈X连锁隐性遗传;病情进行性加重。
如果您还有什么不明白的,请咨询在线医生或拨打咨询热线400-7078-333。



























 冀公网安备 13010402001899号
冀公网安备 13010402001899号